Critical Instruments and Services using a compliant process based approach

ABSTRACT:
The automation industry has served customers in the ‘regulated industries’ which include Food and Beverages, Fine Chemicals, Pharmaceuticals, Biotechnology, Life Sciences, Cosmetics, delivering process measuring instruments, services and solutions dedicated to the processes of these regulated industries: flow, level, pressure, temperature, analysis, (conductivity, pH, OD, …) and recorders.
From this encompassing product range, tailored solutions combined together with services can be delivered as process automation packages, to be used in existing plants and for new projects. Process solutions are implemented together with end users and third parties, for whom good practices are of vital importance, ensuring the best possible quality for the production of the drugs.
FOCUS ON ALL THE REGULATED INDUSTRIES AND KEY REGULATORY REQUIREMENTS:
Regulated industries have overlapping areas and are driven by many of the same laws, rules and recommendations. The FDA’s regulations will become more and more stringent in the future, and the rules and laws have to be well understood and implemented by the end users as well as by the suppliers.
For this reason the automation suppliers have implemented a compliant strategy, to ensure that products and solutions provide the best possible contribution to the final quality of the manufactured drugs. Achieving this objective means having the ability to deliver compliant packages, instrumentation and measuring solutions, including the associated services. The roadmap of Validation is described in a Corporate Validation Master Plan, justifying the strategy and outlining many relevant aspects including documentation, planning, GxP’s areas, GxP’s relevant systems, internal rules, trainings, planned activities.
A COMMON UNDERSTANDING FOR ALL THE PARTNERS: GXP’S, GAMP4, PART 11:
The Validation roadmap must ensure policies and procedures comply with the Good Practices developed by the industry. Regarding measurement instruments, the key regulatory requirements are typically those described in the GAMP Good Practice Guide – Calibration Management published by the ISPE 1 January 2002.
Additionally the GAMP4 2 guidance allows companies to achieve validated and compliant automated solutions meeting all current healthcare regulatory expectations, by building upon existing industry good practice in an efficient and effective way. It allows all parties involved to speak the same language and to have a common understanding.
In August 1997, the title 21 of the Code of Federal Regulation Part 11 became effective. It concerns electronic records and signatures. This law is intended to facilitate the use of paperless record keeping systems and provides criteria under which the FDA will consider electronic records to be equivalent to paper records and electronic signatures to be equivalent to handwritten signatures.
The final rule as stated in section 11.1(a) provides “criteria under which the agency considers electronic records, electronic signatures, and handwritten signatures executed to electronic records to be trustworthy, reliable and generally equivalent to paper records and handwritten signatures executed on paper”.
This law is executed in two parts: electronic records as described in the subpart B of the part 11 and the electronic signatures described in the subpart C of the part 11.
A specific interpretation of 21 CFR Part 11 should always be considered within the Validation Master Plan in line with the scope of supply.
MORE AWARENESS….LESS MISTAKES:
The objective is to produce drugs of the highest quality. A critical instrument produced on a compliant process-based approach is a must for all the regulated industries to achieve this target. More awareness on this issue avoids mistakes and provides many benefits:
- Compliance for the end user
- Knowledge benefits:
- – better understanding of the process
- – improved operational efficiency
- – reduced risks of failure
- – maintenance of quality standards
- Business benefits of the Good Practices:
- – reduced operating costs
- – improved profitability
GLOBAL COMPLIANCE FOR GLOBAL BUSINESS AND GLOBAL CUSTOMERS:
According to IMS Health, the world’s Top 20 pharmaceutical companies are responsible for roughly 65% of the global production of drugs. This clearly demonstrates that the business is highly global, and consequently the key suppliers must be represented on a worldwide basis and be able to demonstrate global compliance. This is the only way, with extensive efforts, extended internal communication and networking, to accumulate experiences, partnerships, exchanges and improved awareness. All of which are needed to serve global customers properly.
COMPLIANT HARDWARE AND SOFTWARE, BUT ALSO SKILLED PEOPLE AND QUALIFIED SERVICES:
Products, Services and Solutions must be compliant, according to the rules. But the people involved in the business are of equal importance. All Staff supporting the regulated industries must be trained on the Good Practices, this includes Management, Marketing personnel, Sales, Project Teams, Sales engineers, Service staff, Documentation specialists, etc. . . Projects should be quoted according to the Good Practices guidelines by the Sales organisations whilst the products must be assembled and tested accordingly to the relevant SOP’s in the Production Centers.
Specific training sessions regarding GxP and in particular 21 CFR part 11 compliance are organised in both production and sales centers and for our IT specialists. Each GxP relevant area has its own action plan and company specific VMPs are implemented where appropriate within the individual countries. Regular on site regular verification and calibration are conducted by qualified service staff trained on Good Practice.
FROM R&D TO . . . DECOMMISSIONING:
Deliverables should be approved in a logical order. A complete life cycle covers the phases of planning, specification, design, construction, testing, installation, acceptance and operation right through to decomissionning of the plant.
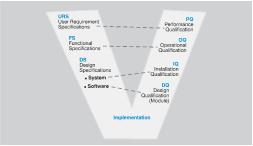
The User Requirement Specifications (URS), the Functional Specifications (FS) along with all the different ‘V-model’ steps are established qualification milestones providing standardization throughout the regulated industries.
The great benefit of this standardization is that it provides a common language, terminology and a comprehensive flow of procedures and tasks which are essential for the implementation of projects and subsequent maintenance.
At the end of its operational life, any system or device should be decommissioned, which may include archiving data. Regulatory or other requirements for the preservation of electronic records should be carefully considered. An archive report should be generated describing the archive approach and listing documents, raw data and electronic records archived. It should be able to retrieve the data even if the original hardware is not anymore available. This data should be retained as required by relevant regulation and user company policy.
IMPLEMENTATION OF THE LATEST TECHNOLOGIES:
The latest Fieldbus digital transmission technologies (Profibus for example) improve substantially the integrity of measurement, increase instrument performance and at the same time help to simplify the documentation. Consequently operating costs can be reduced. It is possible to provide compliant Engineering tasks and documents regarding Fieldbus technologies as well as solutions and tools for qualification and monitoring of sensors and systems using digital technologies.
Many vendors are actively engaged in the PNO group, pushing and developing guidelines and references in order to get the best possible procedures and qualification tools for digital technologies.
DOCUMENTATION . . . AND DOCUMENTATION MANAGEMENT:
Ask the following question to a drugs manufacturer “What’s your first concern?” He will probably answer “To get a compliant production tool . . . This is my first target!” And even when a production unit is compliant, everything must be documented. This is also the same situation with any product, solution or service: “if it is not documented, it is not done”. Documentation is a major part of global compliance.
The purpose of Good Documentation Practice is to ensure that key documents are created, reviewed, approved, distributed and stored in a controlled manner. This should ensure that key documents, such as requirement documents, design documents, as-built documents, tests results, calibration records, etc . . . are used properly and established in a traceable and manageable basis for qualification and validation activities. As a compliant supplier we recognise and support this need from the project design stage right through to provision of tools for calibration management.
OBSERVATIONS AND CONCLUSIONS:
It is imperative to understand the manufacturers business, to conceive, design, produce, test and deliver measurement devices, services and solutions with the appropriate features to adequately meet pharmaceutical and regulated industry expectations.
To be able to rely on a partner having accumulated experience, flexibility, understanding, know how and partnership mentality is of premium importance.
Global approach, costs benefits, mutual comprehension, product quality, increased safety and compliance, trained people are the key areas where added value is created, time is saved, and knowledge is developed for all the parties.
The only way to achieve this goal is to implement a compliant processed approach, based on the Good Practices guidelines. This is something like a ‘universal and profitable language’.
Acronyms
- FDA: Food & Drug Administration
- ISPE: International Society for Pharmacoepidemiology
- GxP’s: Good manufacturing Practises
- GAMP: Good Automated Manufacturing Practise
- PNO: Profibus Nutzeorganisation (usergroup)
- SOP: Standard Operating Procedures
- ISPE, GAMP Good Manufacturing Practise Guide : Calibration management
- ISPE, GAMP4 Good Automated Manufacturing Practise
- 2 1 CFR Part 11 – Electronic Records and Signatures Section 11-10(e). Department of Food and Drug Administration.